The User Guide of Ligand-Based Virtual Screening
1. Introduction to LBDD
Accurate prediction of the interaction between small drug molecule and target protein is the key in virtual screening for drug discovery (VS). Ligand-Based Drug Discovery (LBDD) is a computational technique used in VS. Through searching libraries of small molecules structures, LBDD learn to model the structure-activity relationships, and predicts the biological activity of new compounds in various assays.
The number of tested compounds for a given target is often insufficient to learn a robust structure-activity. To solve this problem, our platform introduces the meta-learning algorithms for the LBDD task. We are the first to predict the relationships with deep neural networks which learns a set of initialized parameters for the deep neural network based on ChEMBL activity data. This approach only requires a small amount of ligand activity data to predict accurately on the target. The deep neural network is modularized, allowing each receptor develops a specified pathway strategy for selecting related modules. In this design, receptors corresponding to the same pathway are considered to be highly similar. The high similarity of the ligands learned by this algorithm also guides the drug molecular design.
Note: Assay IDs used on this platform are consistent with the ChEMBL Assay IDs in the ChEMBL database.
2. Start to Use
2.1 Select Assays (required)
We provide two methods to select assay IDs. You may select relevant assay IDs through targets, or specify the assay ID directly.
-
1) Select Assays through Targets
As shown in the figure below, you can input a target in the input box, then click "Submit". An assay ID list would pop up and you can tick off the relevant assay IDs you need.
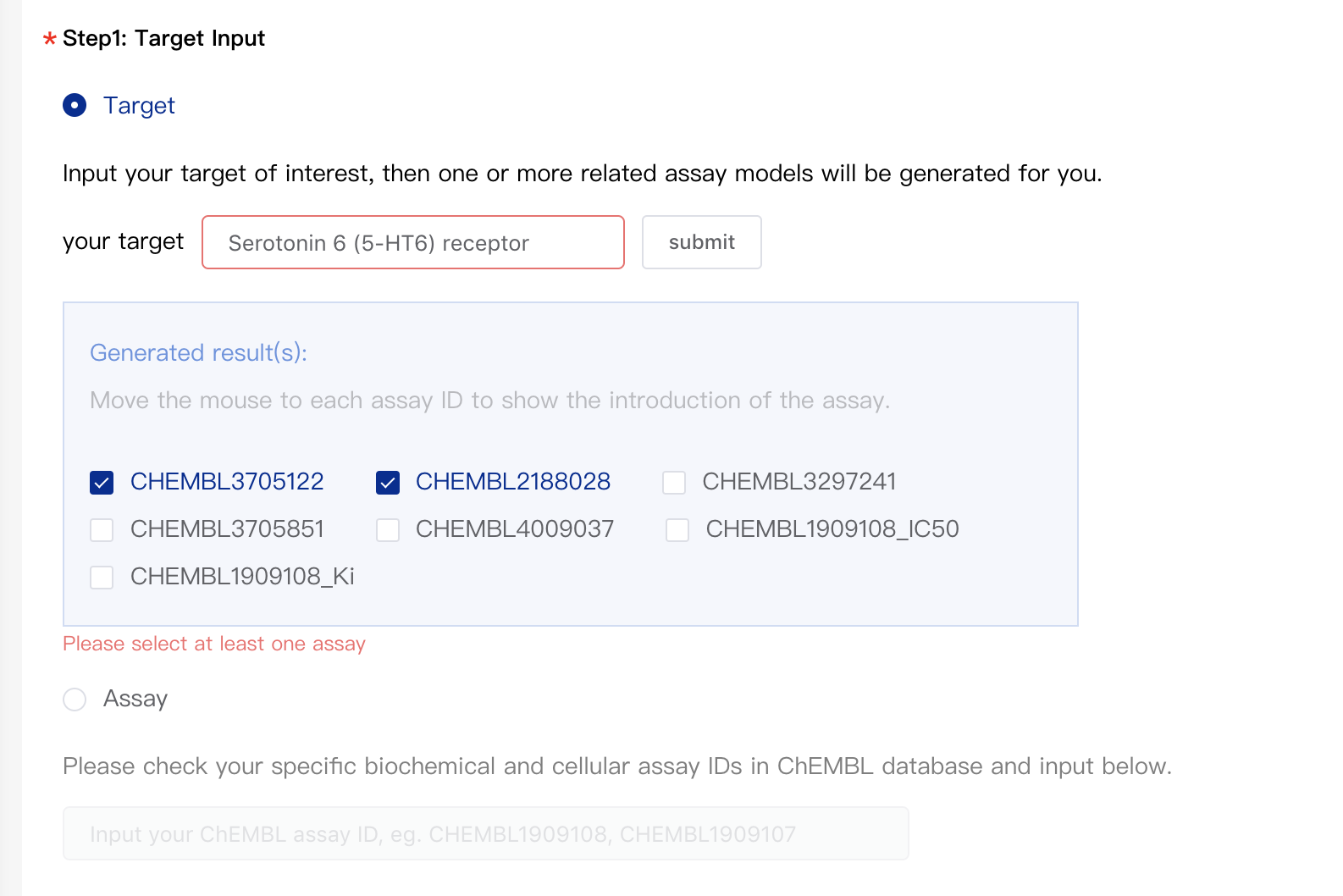
-
2) Specify Assay IDs Directly
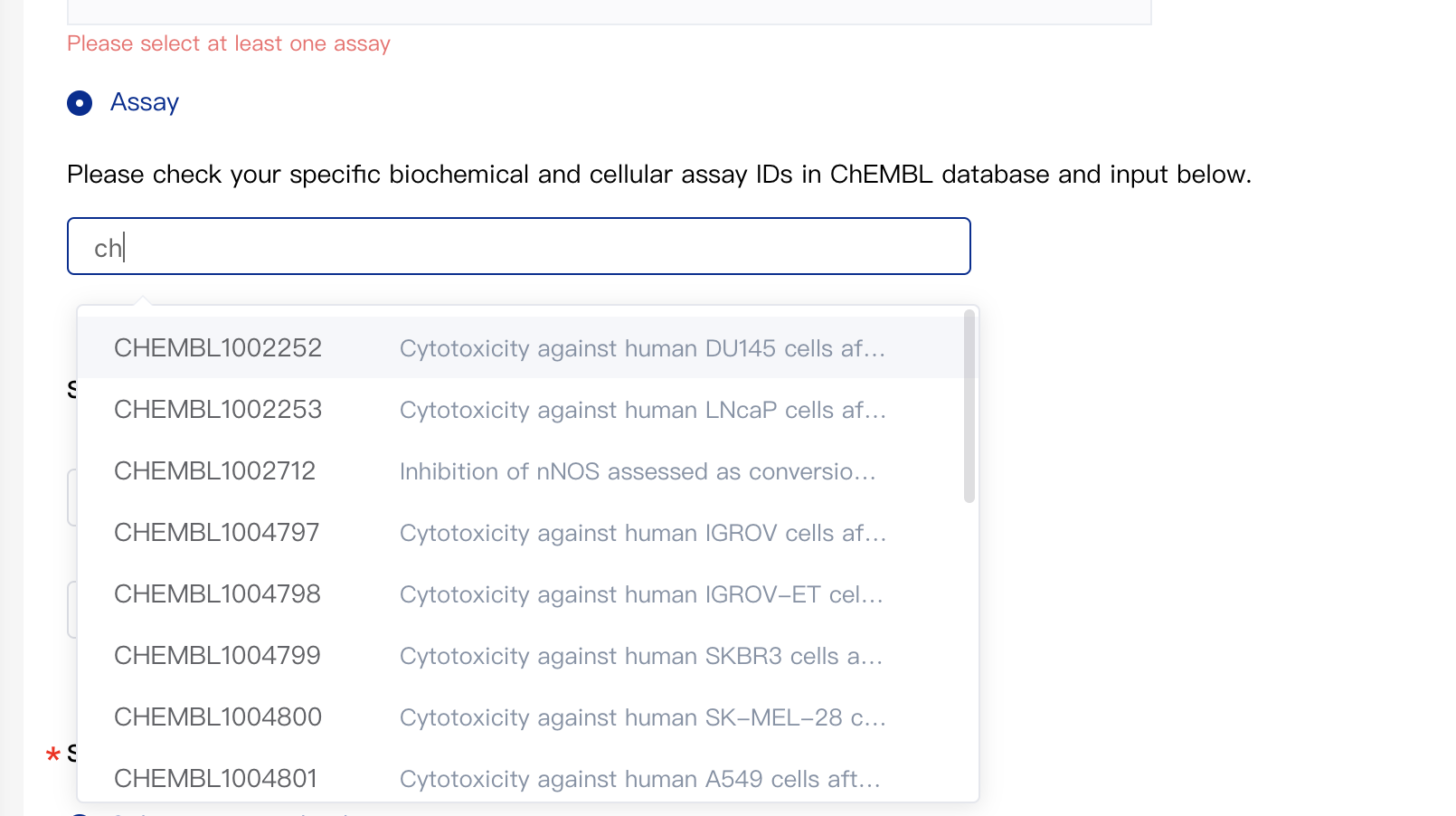
If you've already known your assay IDs or the assay list needed is not corresponding to a single target, then you may select assay IDs by specifying them directly. Please click "Assay" and then input assay IDs in the input box below. The platform will automatically suggest possible IDs based on what the user has entered.
2.2 Select Safety Panel Assay (Optional)
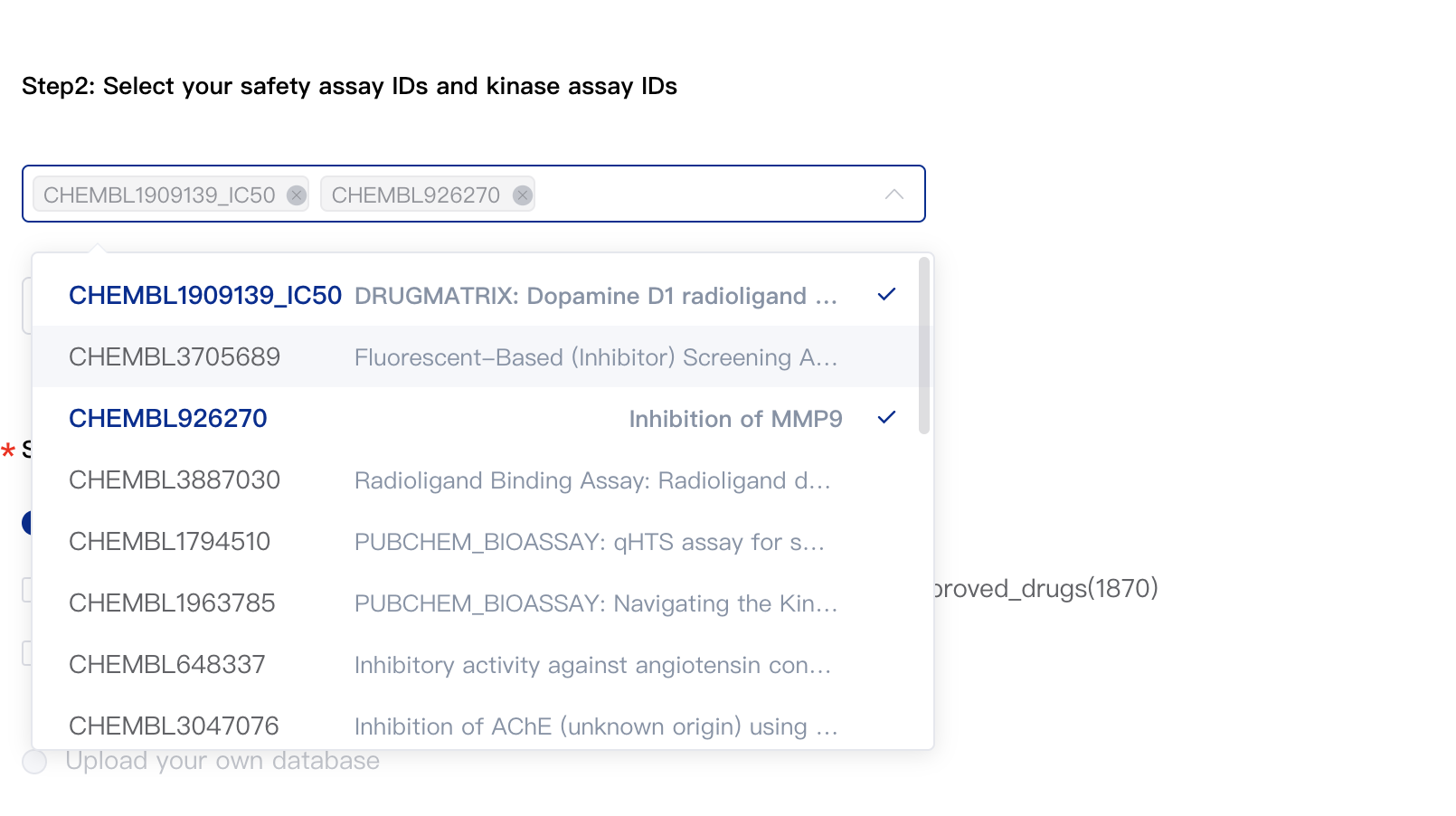
The platform provides 34 assays related to drug safety. You can specify the assays you need by assay IDs.
2.3 Select Kinase Assay (Optional)
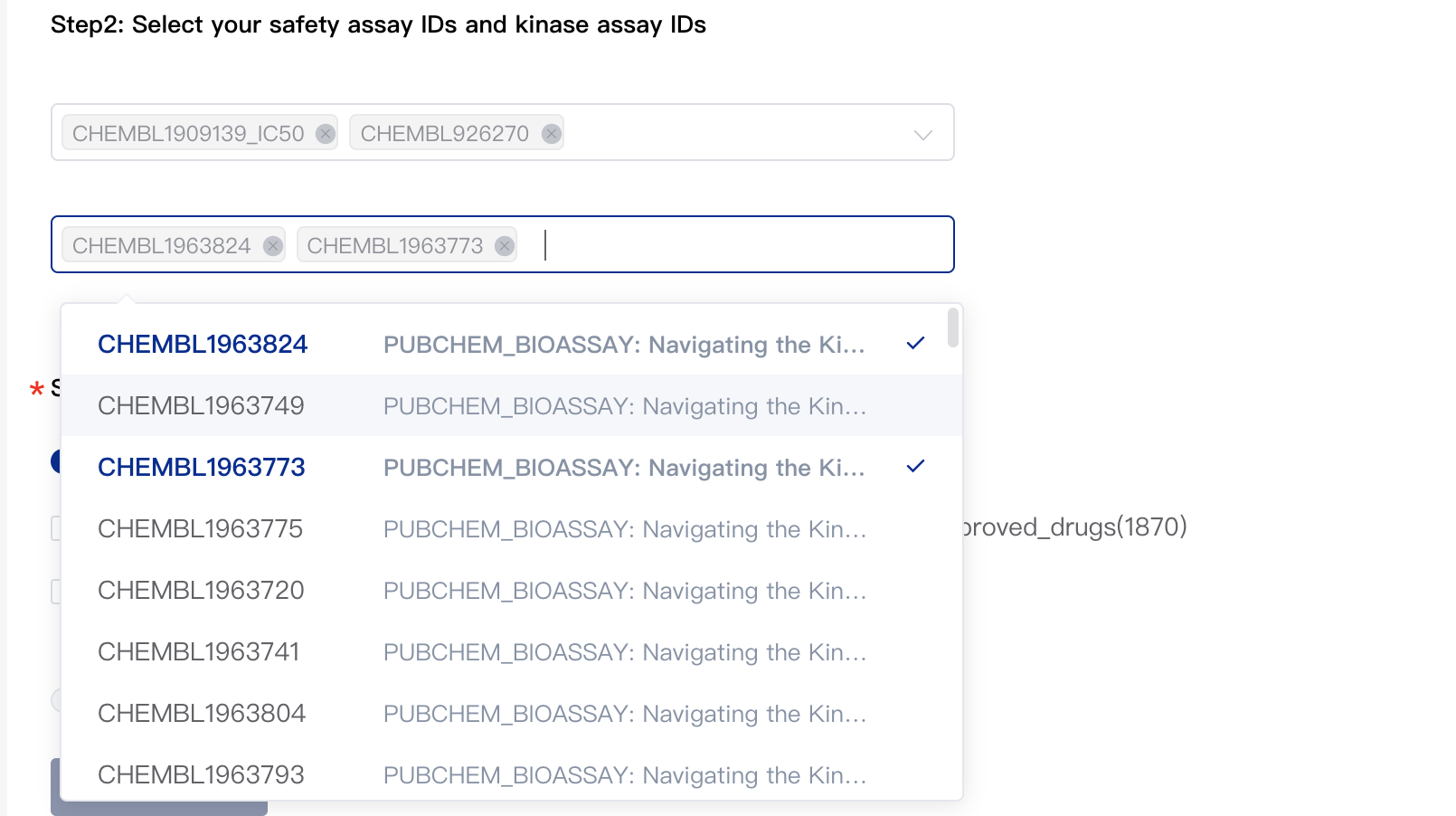
The platform provides 99 assays related to kinase selectivity. You can specify the assays you need by assay IDs.
2.4 Select Compound libraries
You may select one or more databases from 11 public databases the platform has integrated. Activity prediction data on all assays will be provided according to your selected library or libraries.

2.5 Submit Your Task
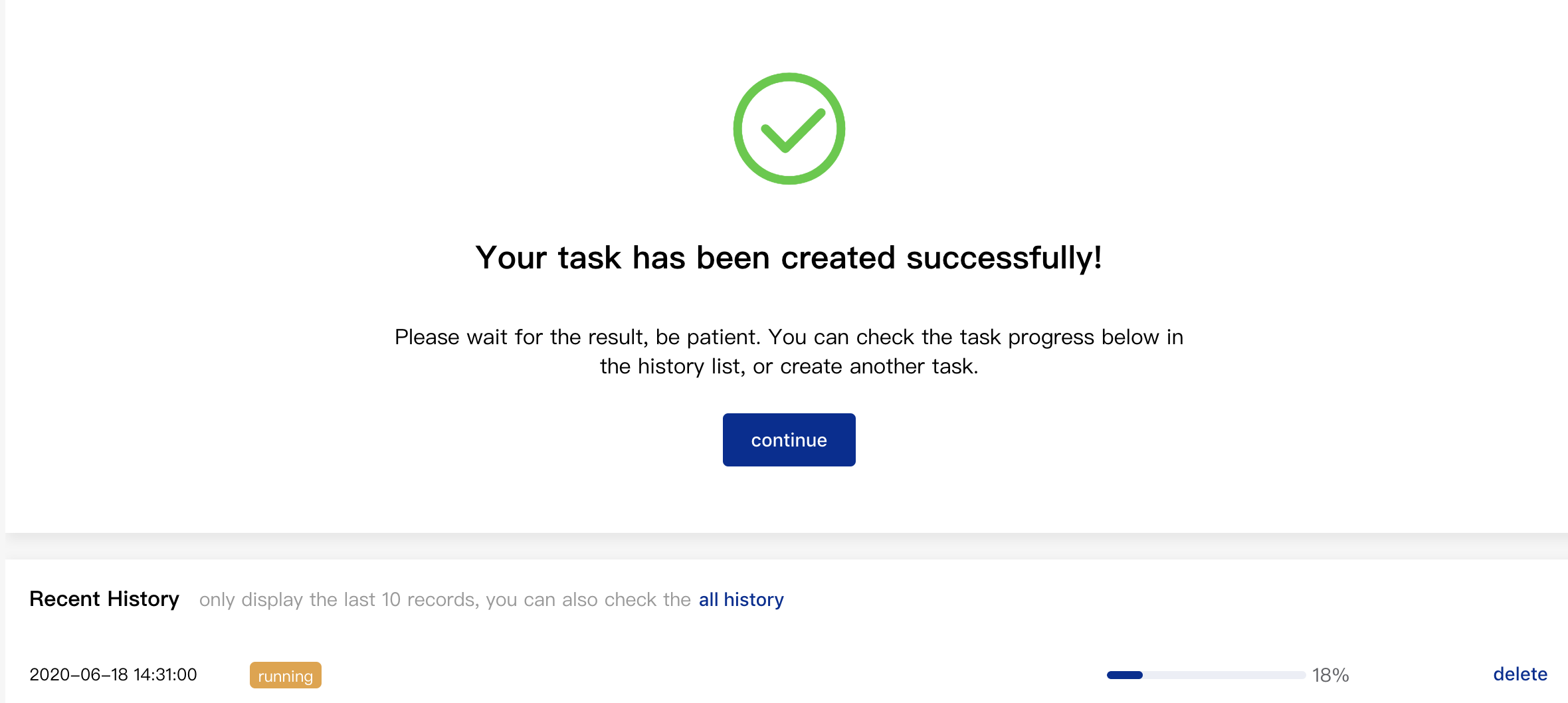
Once the parameters above are set, please click "Submit" to submit your task. The page below shows your submission has succeeded. You can click "Recent History" below to check the processing progress or delete your operations.
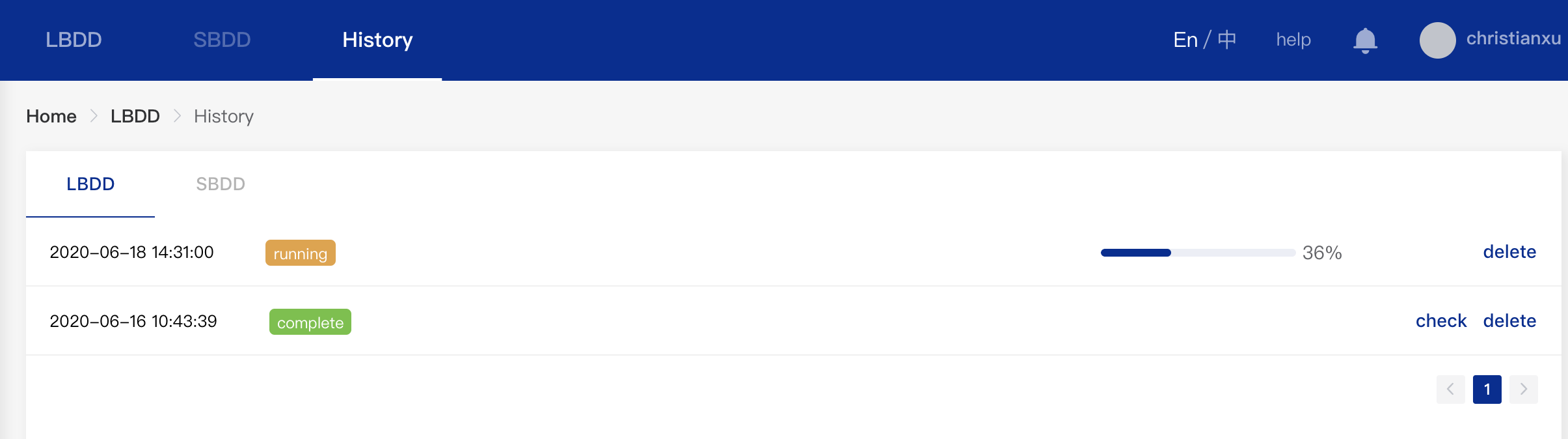
2.6 Query the Result of Your Task
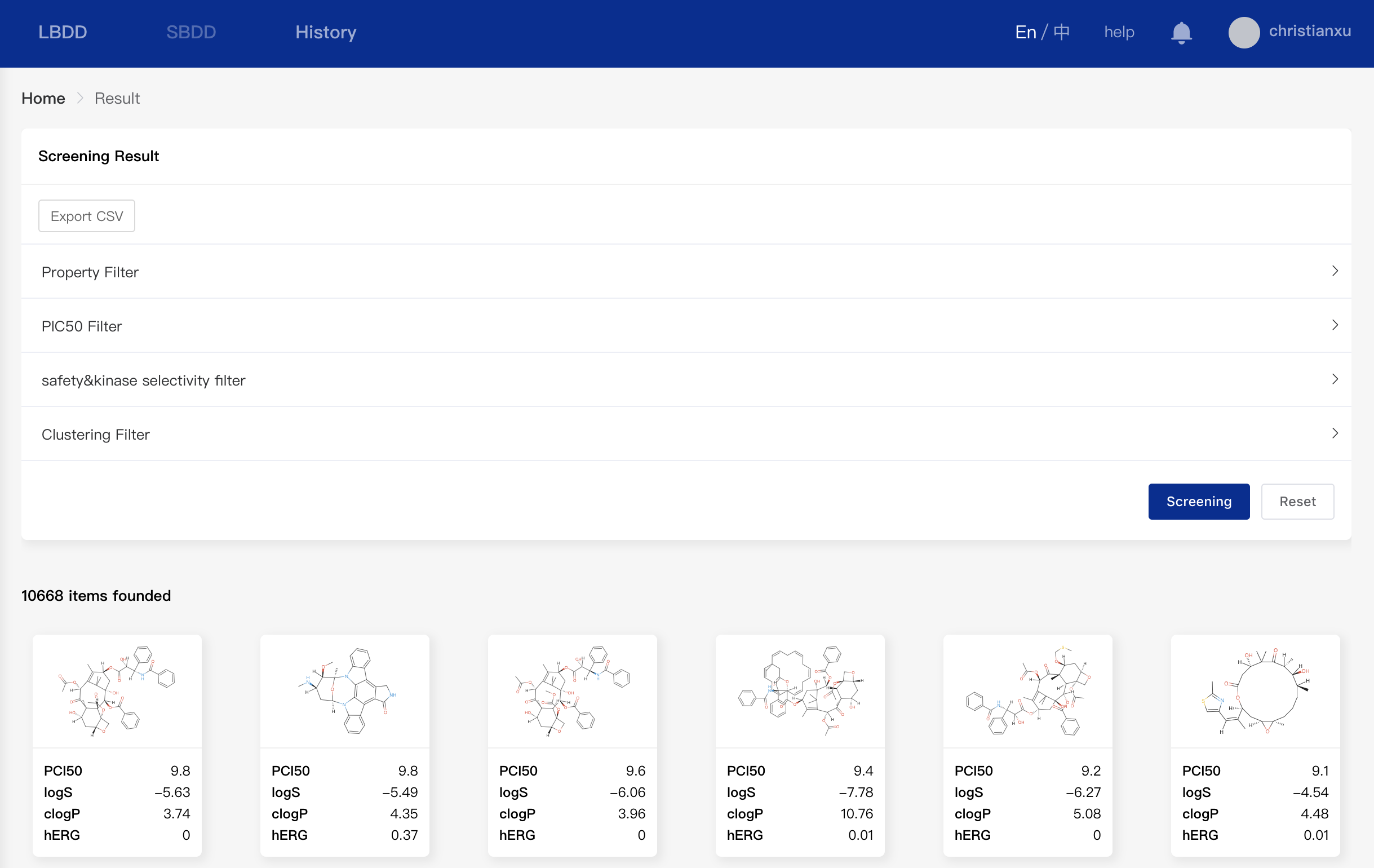
You can query the results of your past tasks by clicking "History" on the top. As shown below, you can filter the results by setting relevant thresholds such as physicochemical property thresholds, pIC50 thresholds, pIC50 thresholds related to safety & kinase selectivity, and clustering filter. The filtering result will be exported to a CSV file.
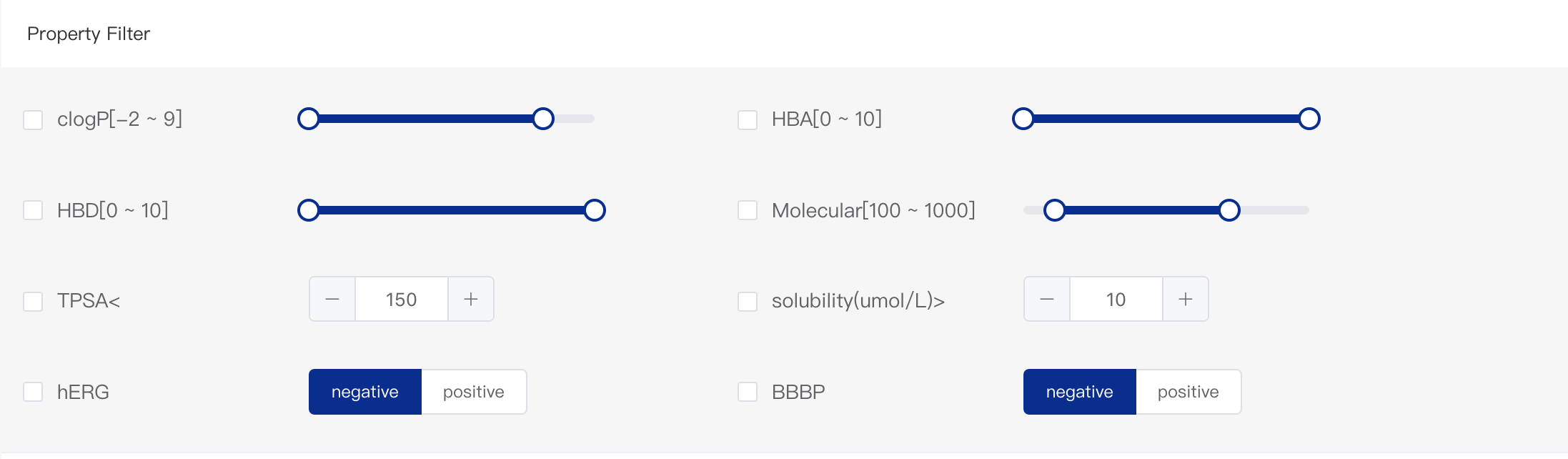
- Physicochemical Properties Filter
There are eight different properties that can be adjusted for result filtering, including basic molecular properties and ADMET properties. They are lipid-water partition coefficient (logP), hydrogen bond acceptor (HBA),hydrogen bond donor (HBD), molecular weight, topological polar surface area (TPSA), solubility (in μmol/L), hERG toxicity ("negative" means the predicted toxicity probability is lower than 0.3 and "positive" means the predicted toxicity probability is larger than 0.3), and blood-brain barrier permeation rate (BBBP; "negative" means the probability is lower than 0.5 and "positive" means the probability is larger than 0.5).
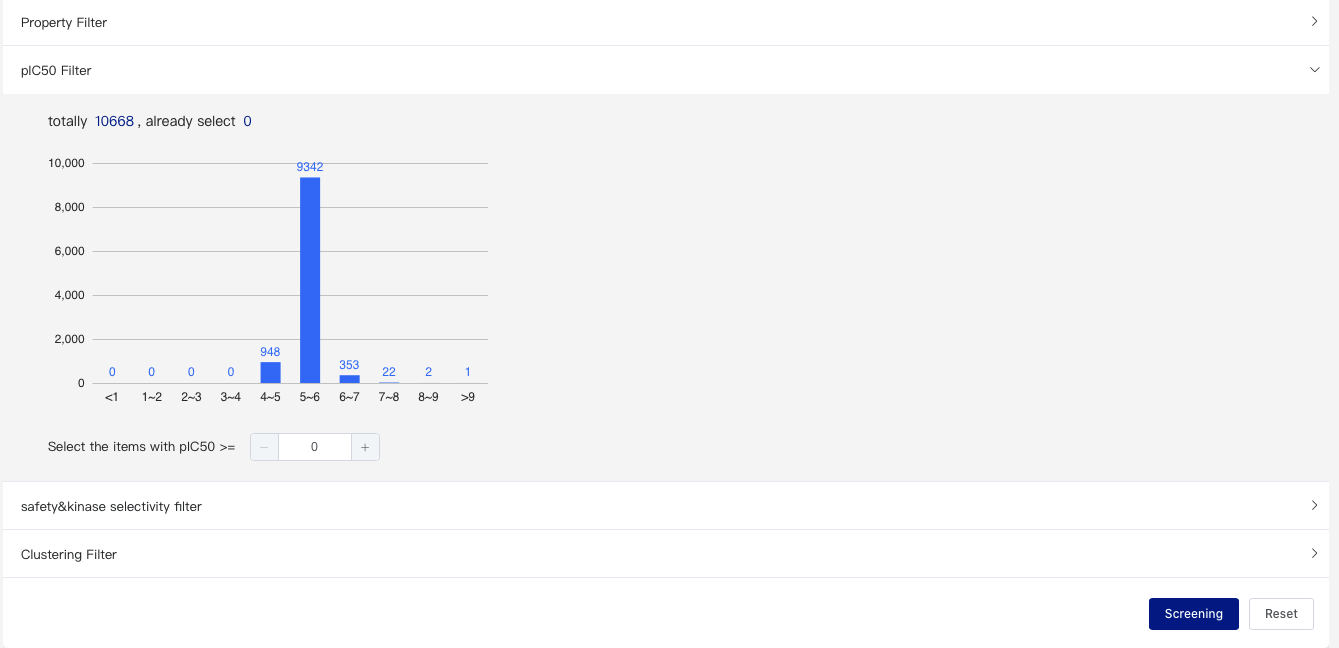
- pIC50 Filter
You can set a filtering range on pIC50 to select molecules for subsequent screening. The pIC50 value here is the predicted activity for the assay you selected in the step 1 above. If multiple assays were selected, the pIC50 value here would be the average of the pIC50 values of them.
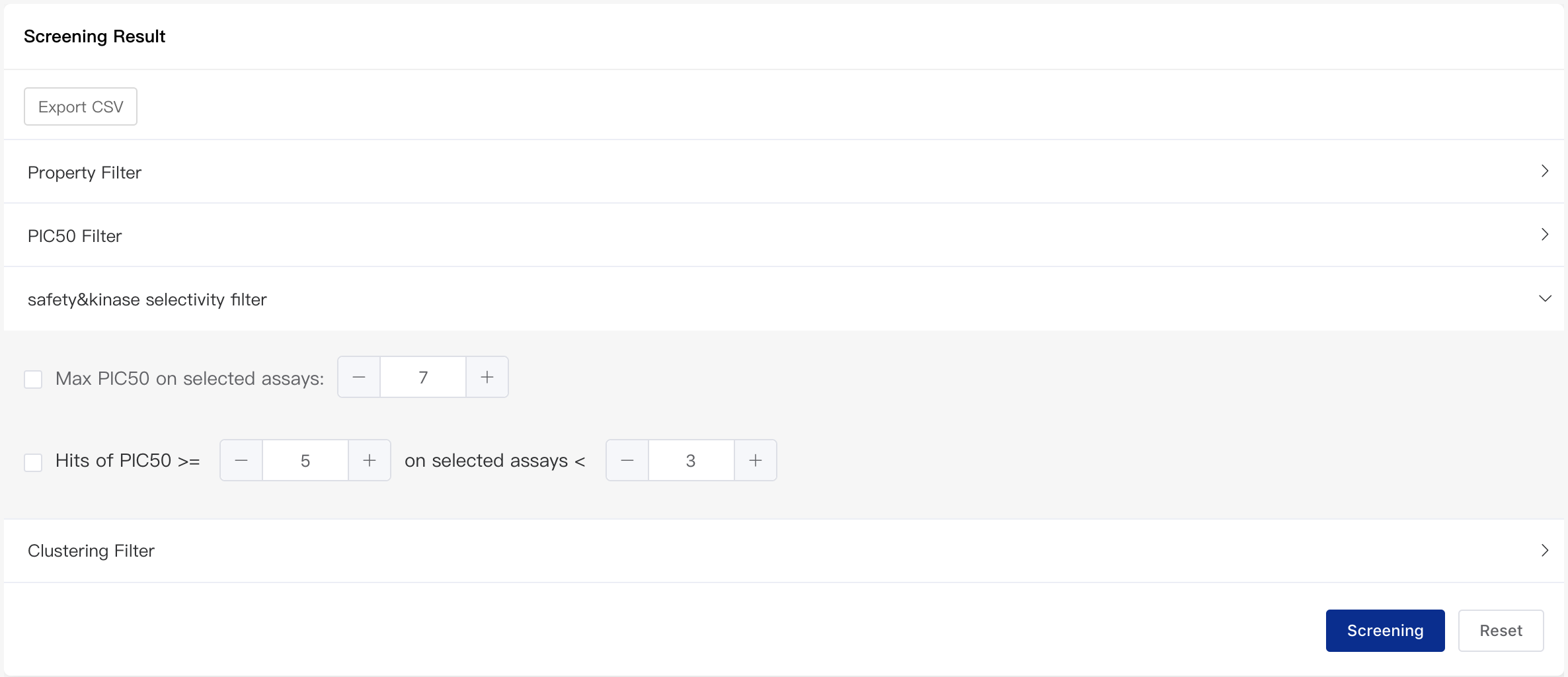
- Safety & Kinase Selectivity Filter
The platform supports selecting safety panel assay and kinase assay when predicting activity as described above. This is mainly to incorporate consideration of off-target toxicity. You can specify a maximum value for the predicted activity of the selected assays. You can also specify a maximum number for the assays that have a certain predicted activity. For example, you can limit the pIC50 for the selected assay to no more than 7 (this value represents obvious toxicity) or you can limit the number of selected assays that have a pIC50 larger than 5 to no more than 3.
- Clustering Filter
The Tanimoto similarity of the molecules will be calculated.Molecules having a similarity larger than 0.6 will be clustered to one cluster. You can specify the number of molecules you want to select from each cluster.
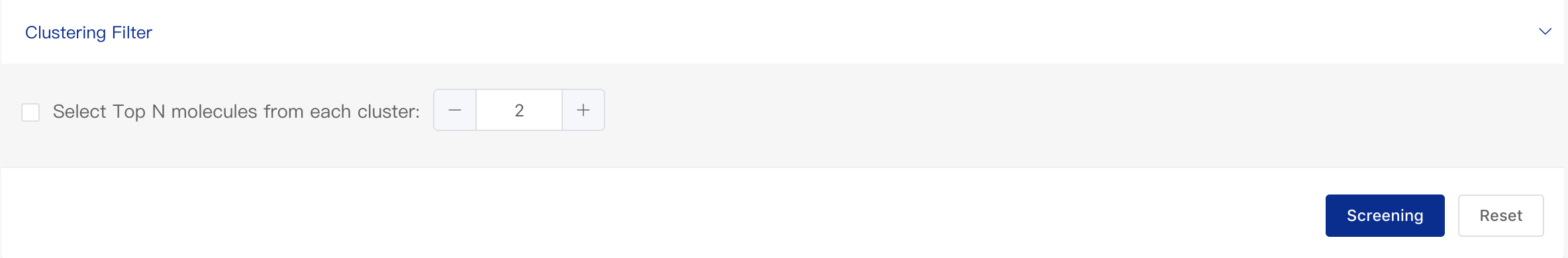
Please click "Screening" to obtain the number of molecules filtered according to the above rules. You can also export the molecules to a CSV file.